After the data has been combined and the reads trimmed to remove adapter contamination has to be aligned, in my case, to a reference genome.
The reference genome is in FASTA format and I usually change the names of the scaffolds or the chromosomes (if they are defined). I also edit the gff3 file that contains the gene annotation for further analysis.
Here there is basic information about alignment and a list of many alignment methods I could try:
https://en.wikibooks.org/wiki/Next_Generation_Sequencing_%28NGS%29/Alignment
Why I'm trying different methods? because using the following method I got a 90% coverage when I was expecting around 95%. Also there is a big difference in the results I obtained from the TruSeq samples than the Nextera. The TruSeq samples contain much more structural variation. Why is that?
The first method I tried is "speedseq":
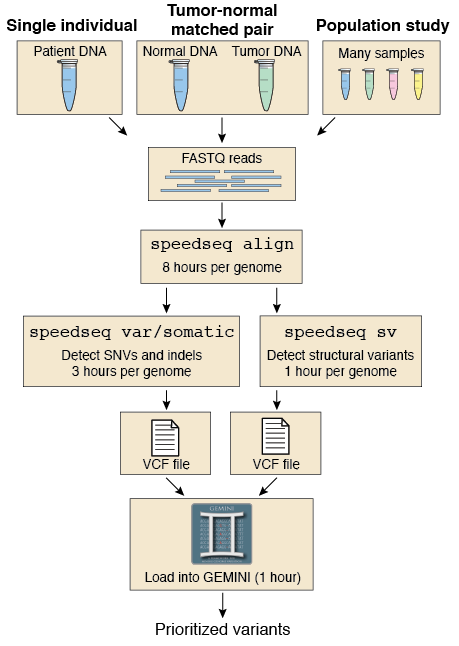 In my case, because my genomes are very small, it takes around 20 min/sample.
In my case, because my genomes are very small, it takes around 20 min/sample.
For some reason, it gives very different alignment coverage results for the 6 samples (data mentioned in my previous post).
It uses BWA mem to do the alignment and a combination of many other tools to get different results:
https://github.com/hall-lab/speedseq/blob/master/example/run_speedseq.sh
The resulting files after running it are:
I have two files but I read this in its documentation:
IMPORTANT: NextGenMap does not check/compare read names. It assumes that the reads are in the correct order. Thus, the input file must only contain pairs of reads. Mixing single end and paired end reads is not supported and will corrupt the mapping results.
Because I am not sure of the order of the reads in the files I tried interleave the files:
The reference genome is in FASTA format and I usually change the names of the scaffolds or the chromosomes (if they are defined). I also edit the gff3 file that contains the gene annotation for further analysis.
Here there is basic information about alignment and a list of many alignment methods I could try:
https://en.wikibooks.org/wiki/Next_Generation_Sequencing_%28NGS%29/Alignment
Why I'm trying different methods? because using the following method I got a 90% coverage when I was expecting around 95%. Also there is a big difference in the results I obtained from the TruSeq samples than the Nextera. The TruSeq samples contain much more structural variation. Why is that?
The first method I tried is "speedseq":
For some reason, it gives very different alignment coverage results for the 6 samples (data mentioned in my previous post).
It uses BWA mem to do the alignment and a combination of many other tools to get different results:
- BWA (http://bio-bwa.sourceforge.net/)
- FreeBayes (https://github.com/ekg/freebayes)
- LUMPY (https://github.com/arq5x/lumpy-sv)
- Sambamba (http://lomereiter.github.io/sambamba/)
- SAMBLASTER (https://github.com/GregoryFaust/samblaster)
- Vawk (https://github.com/cc2qe/vawk)
- GNU Parallel (http://www.gnu.org/software/parallel/)
- mbuffer (http://www.maier-komor.de/mbuffer.html)
https://github.com/hall-lab/speedseq/blob/master/example/run_speedseq.sh
The resulting files after running it are:
- example.bam
- example.discordants.bam
- example.splitters.bam
- example.vcf.gz
- example.sv.vcf.gz
I have two files but I read this in its documentation:
IMPORTANT: NextGenMap does not check/compare read names. It assumes that the reads are in the correct order. Thus, the input file must only contain pairs of reads. Mixing single end and paired end reads is not supported and will corrupt the mapping results.
Because I am not sure of the order of the reads in the files I tried interleave the files:
ngm-utils interleave -1 forward_reads.fq -2 reverse_reads.fq \
-o suffled_reads.fqThe benefit of using ngm-utils interleave is that the read names of the
pairs are checked. If there are reads without a mate present, they will
be filtered.
It produces a sam file but I want the same output than with "speedseq" so I use this command from "samtools" to transform the sam file into bam file:samtools view -bT $ref $sam > $outputThe next script produces all the needed output files from a bam file (this script is not mine but now I can't find the place from where I copied). It uses samblaster to do it like speedseq does.#!/bin/bash BAM=$1 OUT_DIR=$2 LUMPY_HOME=src/lumpy-sv SAMBAMBA=bin/sambamba SAMBLASTER=bin/samblaster BAMLIBS=$LUMPY_HOME/scripts/bamkit/bamlibs.py SAMTOBAM="$SAMBAMBA view -S -f bam -l 0" SAMSORT="$SAMBAMBA sort -m 1G --tmpdir " PAIREND_DISTRO=$LUMPY_HOME/scripts/pairend_distro.py BAMGROUPREADS=$LUMPY_HOME/scripts/bamkit/bamgroupreads.py BAMFILTERRG=$LUMPY_HOME/scripts/bamkit/bamfilterrg.py MAX_SPLIT_COUNT=2 MIN_NON_OVERLAP=20 if [ ! -d "$OUT_DIR" ]; then mkdir $OUT_DIR fi BAM_BASE=`basename $BAM .bam` OUTPUT=`basename $BAM`.vcf OUTBASE=`basename "$OUTPUT"` # make pipes TEMP_DIR=$OUT_DIR/$BAM_BASE mkdir -p $TEMP_DIR/spl $TEMP_DIR/disc mkfifo $TEMP_DIR/spl_pipe mkfifo $TEMP_DIR/disc_pipe echo "$PYTHON $BAMGROUPREADS \ -i $BAM \ | $SAMBLASTER \ --acceptDupMarks \ --excludeDups \ --addMateTags \ --maxSplitCount $MAX_SPLIT_COUNT \ --minNonOverlap $MIN_NON_OVERLAP \ --splitterFile $TEMP_DIR/spl_pipe \ --discordantFile $TEMP_DIR/disc_pipe " $PYTHON $BAMGROUPREADS \ -i $BAM \ | $SAMBLASTER \ --acceptDupMarks \ --excludeDups \ --addMateTags \ --maxSplitCount $MAX_SPLIT_COUNT \ --minNonOverlap $MIN_NON_OVERLAP \ --splitterFile $TEMP_DIR/spl_pipe \ --discordantFile $TEMP_DIR/disc_pipe \ > /dev/null & $SAMTOBAM $TEMP_DIR/spl_pipe \ | $SAMSORT $TEMP_DIR/spl \ -o $TEMP_DIR/$BAM_BASE.splitters.bam \ /dev/stdin & $SAMTOBAM $TEMP_DIR/disc_pipe \ | $SAMSORT $TEMP_DIR/disc \ -o $TEMP_DIR/$BAM_BASE.discordants.bam \ /dev/stdin wait rm $TEMP_DIR/spl_pipe rm $TEMP_DIR/disc_pipe rmdir $TEMP_DIR/spl rmdir $TEMP_DIR/disc
Using this alignment I don't get any SV for any of the samples. And Interleaving the reads doesn't not change much the results. Why the splitter.bam file is emptyfor all of them? Is it the algorithm filtering them?
No comments:
Post a Comment